
There is a tremendous need for software that identifies new proteins and aberrant protein forms within proteomic data. This is especially true when analyzing cancer proteins wherein malfunctions of cellular processes have been shown to create protein products that do not appear in healthy cells. Unfortunately, current classification of tumor proteins centers on quantifying the presence of known proteins. This ignores what could be the most profound elements: Proteins with amino acid variations, alternative splice forms, new exons, coding from what should be untranslated regions of RNA, and even entirely new genes from regions that, in healthy cells, should not be genic.
Proteogenomics offer huge potential for the discovery of novel protein coding. The identification of disease-specific proteins could lead to valuable biomarkers or drugs that specifically target diseased pathways.
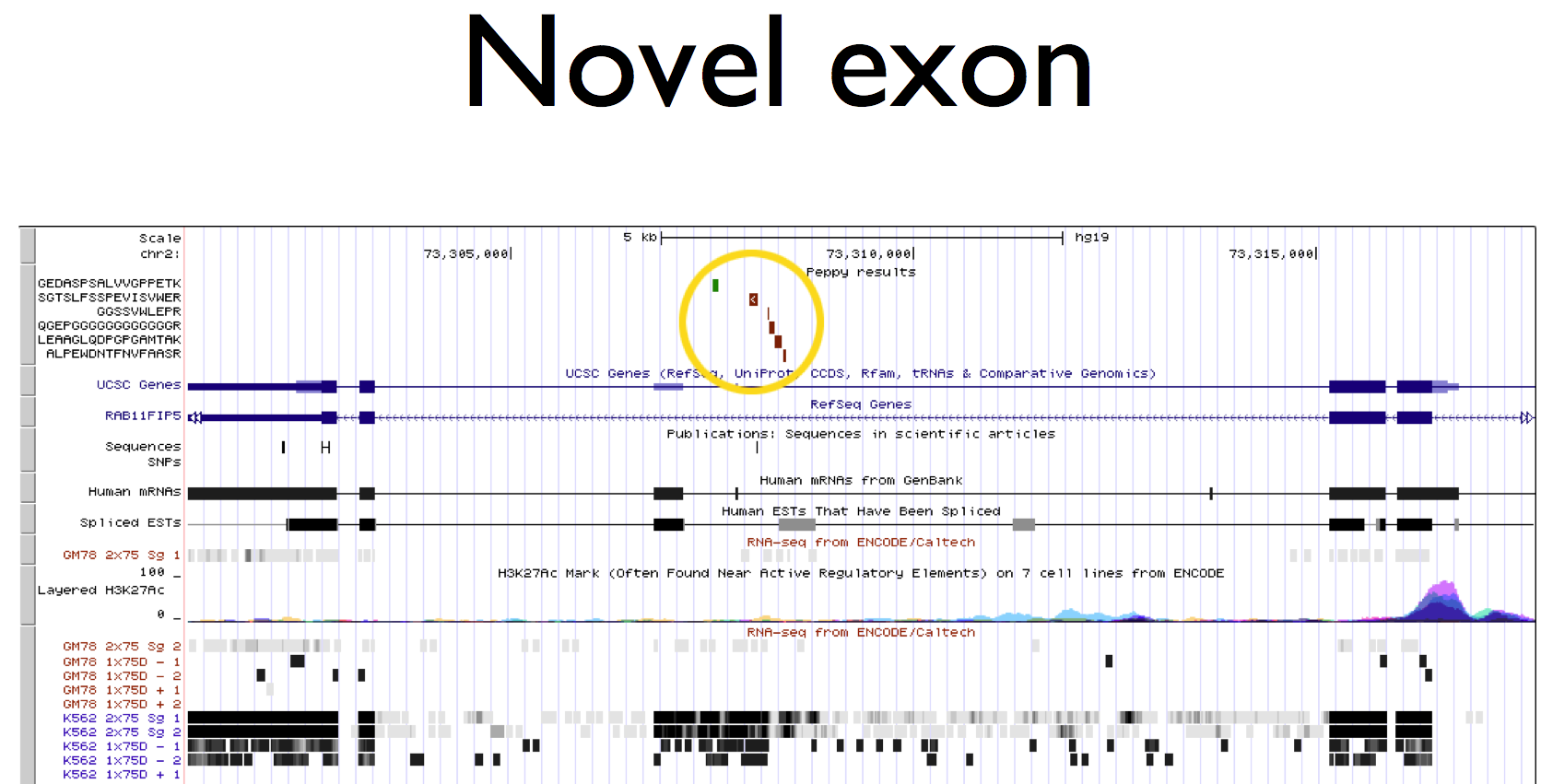
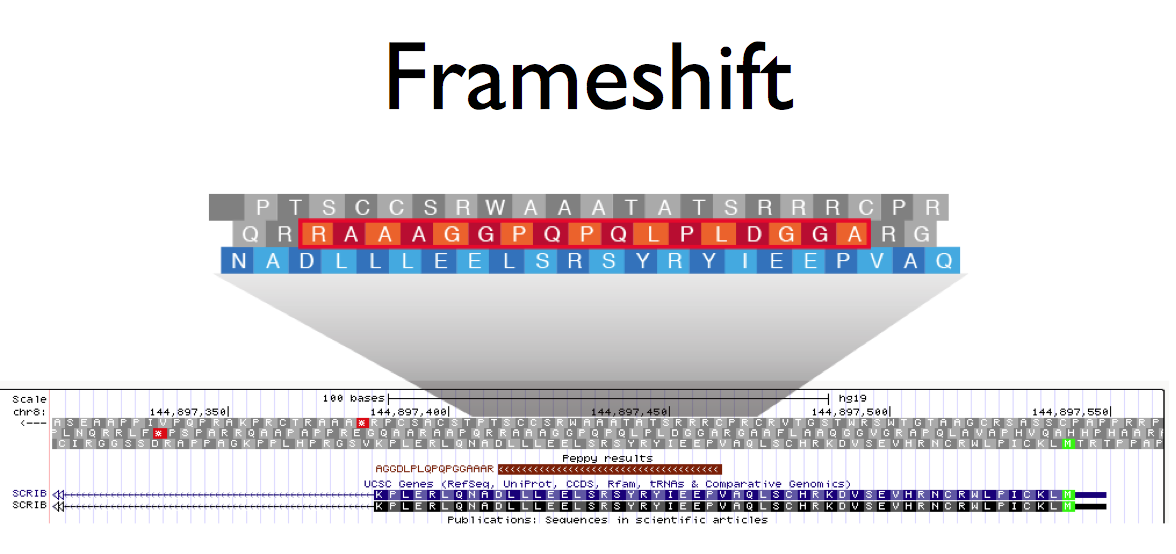
Until now, proteogenomic analysis has been very laborious requiring segmented, 6-frame translations of the genome, results unification and all of the steps for target/decoy false discovery rate.
Peppy automates each of these steps. All that is required is to give the software the proteomic data, give it the genome, and it performs every necessary task.
Sign up to be given a download link to the Peppy software. If you have a specific question, or are interested in collaborating with us for proteogenomic analysis of your proteomic data please contact brian@geneffects.com or on LinkedIn
Leave a Reply